Accessing the Slots of a SpatialMap Object
Tutorial_Accessing_slots_in_SpatialMap_object.Rmd
library(facil)
library(SpatialMap)SpatialMap Structure
Schematic
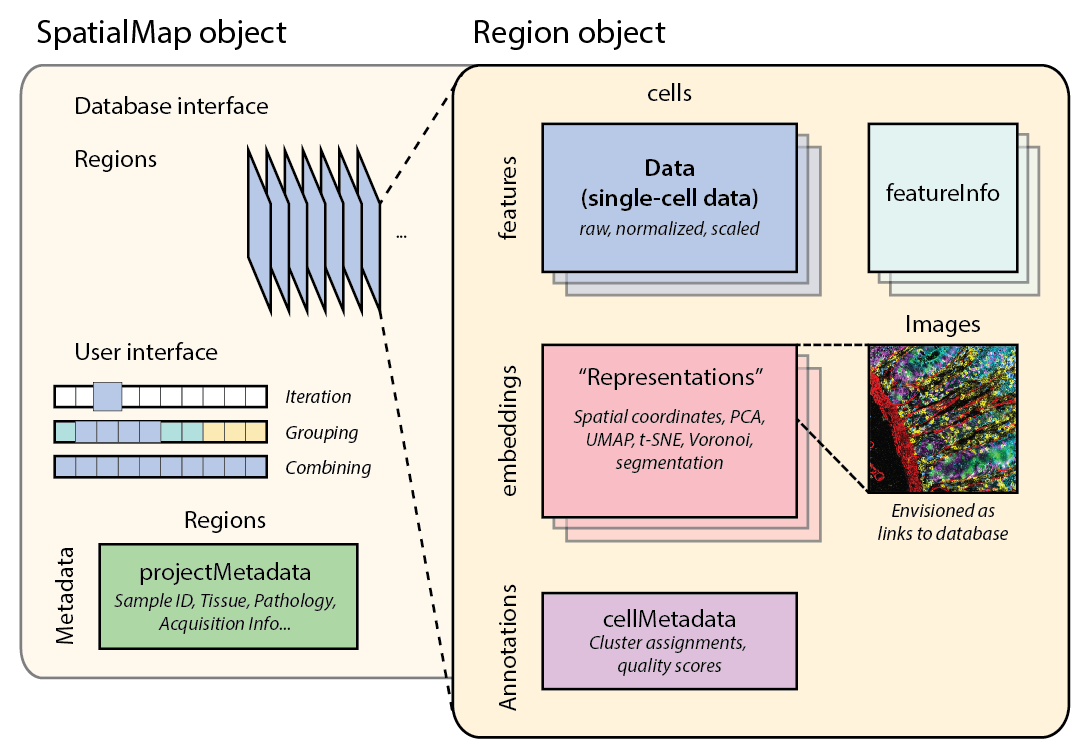 ## R internal schematic
## R internal schematic
To view a SpatialMap (SM) object in R, let’s load one and print the output of str()
sm_tonsil <- load_sm_data("tonsil")
str(sm_tonsil)
#> Formal class 'SpatialMap' [package "SpatialMap"] with 8 slots
#> ..@ regions :List of 2
#> .. ..$ TonsilA:Formal class 'Region' [package "SpatialMap"] with 16 slots
#> .. .. .. ..@ Data : num [1:16, 1:5810] 25.4 616.5 96.7 25.4 192.5 ...
#> .. .. .. .. ..- attr(*, "dimnames")=List of 2
#> .. .. .. .. .. ..$ : chr [1:16] "CD3e" "CD21" "CD68" "Actin" ...
#> .. .. .. .. .. ..$ : chr [1:5810] "TonsilA.125182" "TonsilA.125238" "TonsilA.125254" "TonsilA.125325" ...
#> .. .. .. ..@ NormalizedData : NULL
#> .. .. .. ..@ ScaledData : NULL
#> .. .. .. ..@ bgData : num [1:16, 1:5810] 2242 2328.1 67.1 1433.4 195.4 ...
#> .. .. .. .. ..- attr(*, "dimnames")=List of 2
#> .. .. .. .. .. ..$ : chr [1:16] "DAPI_ch1_cyc7" "DAPI_ch1_cyc8" "Blank_ch2_cyc8" "Blank_ch3_cyc8" ...
#> .. .. .. .. .. ..$ : chr [1:5810] "TonsilA.125182" "TonsilA.125238" "TonsilA.125254" "TonsilA.125325" ...
#> .. .. .. ..@ coordinates :'data.frame': 5810 obs. of 14 variables:
#> .. .. .. .. ..$ study_id : num [1:5810] 266 266 266 266 266 266 266 266 266 266 ...
#> .. .. .. .. ..$ acquisition_id: chr [1:5810] "EnableShare_c001_v001_r001_reg001" "EnableShare_c001_v001_r001_reg001" "EnableShare_c001_v001_r001_reg001" "EnableShare_c001_v001_r001_reg001" ...
#> .. .. .. .. ..$ cell_id : num [1:5810] 125182 125238 125254 125325 125326 ...
#> .. .. .. .. ..$ region : num [1:5810] 1 1 1 1 1 1 1 1 1 1 ...
#> .. .. .. .. ..$ tile_num : num [1:5810] 1 1 1 1 1 1 1 1 1 1 ...
#> .. .. .. .. ..$ x : num [1:5810] 891 935 34 18 997 ...
#> .. .. .. .. ..$ y : num [1:5810] 1996 1993 1991 1985 1986 ...
#> .. .. .. .. ..$ z : num [1:5810] 0 0 0 0 0 0 0 0 0 0 ...
#> .. .. .. .. ..$ x_tile : num [1:5810] 891 935 34 18 997 ...
#> .. .. .. .. ..$ y_tile : num [1:5810] 1996 1993 1991 1985 1986 ...
#> .. .. .. .. ..$ size : num [1:5810] 484 763 357 417 520 651 358 528 370 193 ...
...A bit complex! To abstract the getting and setting of different data types, the SM package provides simple methods to retrieve data in the most common ways. The general framework for this type of object and methods is described in detail in Hadley Wickam’s excellent book on object-oriented programming in R, particularly the section on S4 classes. This approach simplifies the organization of code, and minimizes the unnecessary duplication of data in memory.
Getting and Setting
Here is a demonstration of many “get” methods for SM objects.
SpatialMap-level data
Retrieving Regions and Analyses
## S4 Methods
# Get a Region by name (returns a Region)
getRegion(sm_tonsil, "TonsilA")
#> Region object
#> ID: TonsilA
#> + 5,810 cells
#> + 16 features
# Get multiple Regions, or named subset of them (returns a list)
getRegions(sm_tonsil)
#> $TonsilA
#> Region object
#> ID: TonsilA
#> + 5,810 cells
#> + 16 features
#>
#> $TonsilB
#> Region object
#> ID: TonsilB
#> + 10,694 cells
#> + 16 features
getRegions(sm_tonsil, c("TonsilA", "TonsilB"))
#> $TonsilA
#> Region object
#> ID: TonsilA
#> + 5,810 cells
#> + 16 features
#>
#> $TonsilB
#> Region object
#> ID: TonsilB
#> + 10,694 cells
#> + 16 features
# List Region IDs
Regions(sm_tonsil)
#> [1] "TonsilA" "TonsilB"
## Base R Methods
# Get Regions by indexing
sm_tonsil[[1]]
#> Region object
#> ID: TonsilA
#> + 5,810 cells
#> + 16 features
sm_tonsil[["TonsilA"]]
#> Region object
#> ID: TonsilA
#> + 5,810 cells
#> + 16 features
sm_tonsil$TonsilA
#> Region object
#> ID: TonsilA
#> + 5,810 cells
#> + 16 features
# Subset SpatialMaps by indexing
sm_tonsil[1:2]
#> SpatialMap object
#> Active analysis: regions
#> + 16,504 total cells
#> + 16 features
#> + 2 Regions:
#> - TonsilA
#> - TonsilB
sm_tonsil[c("TonsilA", "TonsilB")]
#> SpatialMap object
#> Active analysis: regions
#> + 16,504 total cells
#> + 16 features
#> + 2 Regions:
#> - TonsilA
#> - TonsilBEquivalent methods are available to access combined single-cell Analyses
sm_tonsil <- createAnalysis(sm_tonsil)
#> New analysis created.
#> 2 Regions
#> 16504 cells
#> 16 common features
#>
#> Added Analysis `combined` to SpatialMap project 'SpatialMap Project'.
#> Setting as active Analysis
#>
# List Analyses
Analyses(sm_tonsil)
#> [1] "combined"
# Get an Analysis
getAnalysis(sm_tonsil, "combined")
#> Analysis object
#> ID: combined
#> + 16,504 cells
#> + 16 common features
#> + 2 Regions:
#> - TonsilA
#> - TonsilBRetrieving other SM slots
SM data slots can also be accessed with SM methods. To see SM slots, and their allowed data types, use the getSlots function.
To pull up a list of SM slots in an interactive IDE (RStudio), use the @ accessor.
getSlots("SpatialMap")
#> regions analyses activeRegions activeAnalysis
#> "list" "ANY" "ANY" "ANY"
#> projectMetadata projectName projectSummary settings
#> "data.frame" "characterOrNull" "characterOrNull" "list"
sm_tonsil@settings
#> $cores
#> [1] 1
#>
#> $parallel
#> [1] "future"
#>
#> $future.initialized
#> [1] FALSE
#>
#> $show.progress
#> [1] FALSE
#>
#> $Rstudio
#> [1] TRUE
#>
...
settings(sm_tonsil)
#> cores parallel
#> "1" "future"
#> future.initialized show.progress
#> "FALSE" "FALSE"
#> Rstudio python.path
#> "TRUE" "none"
#> python.environment python.method
#> "spatialmap_py" "reticulate"
#> platform local.images
#> "unix" "FALSE"
#> filtration normalization
#> "soft" "log"
#> max_cell.size min_cell.size
#> "1600" "100"
#> max_signal.sum min_signal.sum
...
projectMetadata(sm_tonsil)
#> color width n_channels height projection_visualizer_uuid
#> TonsilA grayscale 9408 4 9072 96e6af5a-f612-4f97-a294-c580dcd41915
#> TonsilB grayscale 9408 4 9072 f0b41218-209e-4946-9a1d-1fb7fb3e7a0d
#> sample_label n_levels assay_id n_cycles
#> TonsilA EnableShare 5 1 8
#> TonsilB EnableShare2 5 1 8
#> base_image_visualizer_uuid experiment_label axes
#> TonsilA 728d42b7-77ea-4319-a48a-7f7340bf182d enableshare TCYX
#> TonsilB 2007da53-7ede-492e-84b6-f2d2ab32a09d enableshare2 TCYX
#> assay_metadata assay_description
#> TonsilA {"image_mpp": 0.377, "num_amp_cycles": 0}
#> TonsilB {"image_mpp": 0.377, "num_amp_cycles": 0}
#> experiment_id visual_quality assay_name region_display_label
#> TonsilA 164 true CODEX EnableShare
#> TonsilB 355 true CODEX EnableShare2
...
activeRegions(sm_tonsil)
#> [1] "TonsilA" "TonsilB"
activeAnalysis(sm_tonsil)
#> [1] "combined"Setting new values for SM slots
To update these slots, we can use “setter” methods
## Project metadata is a data frame
pm <- projectMetadata(sm_tonsil)
pm$RandomData <- LETTERS[c(5, 16)]
projectMetadata(sm_tonsil) <- pm
activeRegions(sm_tonsil) <- c("TonsilA", "TonsilB")
activeAnalysis(sm_tonsil) <- "regions"
activeAnalysis(sm_tonsil)
#> [1] "regions"Remember that updating activeRegions and activeAnalysis will change the behavior of almost all SM functions!
Region-level data
Most SpatialMap functions are designed to operate on SM objects, even if they actually affect data in individual Regions. As an example, we will look at cellMetadata.
## cellMetadata is stored on an individual Region level
head(sm_tonsil$TonsilA@cellMetadata)
#> cell_id cell.id anno316_some_categories anno916_Leiden_v1
#> TonsilA.125182 125182 TonsilA.125182 yay Cluster 2
#> TonsilA.125238 125238 TonsilA.125238 nay Cluster 2
#> TonsilA.125254 125254 TonsilA.125254 yay Cluster 1
#> TonsilA.125325 125325 TonsilA.125325 yay Cluster 1
#> TonsilA.125326 125326 TonsilA.125326 nay Cluster 6
#> TonsilA.125608 125608 TonsilA.125608 nay Cluster 1
#> anno248_test_subset_annotation_324
#> TonsilA.125182 p
#> TonsilA.125238 u
#> TonsilA.125254 t
#> TonsilA.125325 t
#> TonsilA.125326 b
#> TonsilA.125608 y
#> anno1189_uqc_test10_letters_qc_assoc anno1149_uqc_test10_letters
...
## the Getter function works well for an individual Region
cm1 <- cellMetadata(sm_tonsil$TonsilA)
head(cm1)
#> cell_id cell.id anno316_some_categories anno916_Leiden_v1
#> TonsilA.125182 125182 TonsilA.125182 yay Cluster 2
#> TonsilA.125238 125238 TonsilA.125238 nay Cluster 2
#> TonsilA.125254 125254 TonsilA.125254 yay Cluster 1
#> TonsilA.125325 125325 TonsilA.125325 yay Cluster 1
#> TonsilA.125326 125326 TonsilA.125326 nay Cluster 6
#> TonsilA.125608 125608 TonsilA.125608 nay Cluster 1
#> anno248_test_subset_annotation_324
#> TonsilA.125182 p
#> TonsilA.125238 u
#> TonsilA.125254 t
#> TonsilA.125325 t
#> TonsilA.125326 b
#> TonsilA.125608 y
#> anno1189_uqc_test10_letters_qc_assoc anno1149_uqc_test10_letters
...
dim(cm1)
#> [1] 5810 38
## And for the whole SM object
cm.all <- cellMetadata(sm_tonsil)
## The SM function combines all Region@cellMetadata slots into one data.frame
dim(cm.all)
#> [1] 16504 38Similar rules apply to most of the Region slots. There are slightly different rules based on whether output from multiple Regions is concatenated (as in the cellMetadata) case, intersected (in the case of the features() getter, only features in common for all Regions will be returned), or listed (the output is not combined, but instead returned as a list per Region)
activeAnalysis(sm_tonsil) <- "regions"
## Concatenate
Data(sm_tonsil, "Data") %>% med()
#> Matrix dim 16 x 16504
#> TonsilA.125182 TonsilA.125238 TonsilA.125254 TonsilA.125325
#> CD3e 25.430 32.674 47.148 43.724
#> CD21 616.531 264.544 295.633 342.288
#> CD68 96.736 115.676 400.171 376.578
#> Actin 25.351 30.097 68.339 75.369
#> CD45RO 192.475 159.933 597.997 558.926
#> HisH3p 54.665 55.067 53.076 62.578
#> TonsilA.125326 TonsilA.125608
#> CD3e 26.698 27.512
#> CD21 217.321 241.673
#> CD68 206.606 233.141
#> Actin 32.975 46.071
#> CD45RO 293.971 205.307
#> HisH3p 59.348 57.280
cellMetadata(sm_tonsil) %>% med()
#> Data Frame dim 16504 x 38
#> cell_id cell.id anno316_some_categories anno916_Leiden_v1
#> TonsilA.125182 125182 TonsilA.125182 yay Cluster 2
#> TonsilA.125238 125238 TonsilA.125238 nay Cluster 2
#> TonsilA.125254 125254 TonsilA.125254 yay Cluster 1
#> TonsilA.125325 125325 TonsilA.125325 yay Cluster 1
#> TonsilA.125326 125326 TonsilA.125326 nay Cluster 6
#> TonsilA.125608 125608 TonsilA.125608 nay Cluster 1
#> anno248_test_subset_annotation_324
#> TonsilA.125182 p
#> TonsilA.125238 u
#> TonsilA.125254 t
#> TonsilA.125325 t
#> TonsilA.125326 b
#> TonsilA.125608 y
#> anno1189_uqc_test10_letters_qc_assoc
...
cells(sm_tonsil) %>% med()
#> Character vector, length: 16504
#> [1] "TonsilA.125182" "TonsilA.125238" "TonsilA.125254" "TonsilA.125325"
#> [5] "TonsilA.125326" "TonsilA.125608"
## Intersect
bgFeatures(sm_tonsil)
#> [1] "DAPI_ch1_cyc7" "DAPI_ch1_cyc8" "Blank_ch2_cyc8" "Blank_ch3_cyc8"
#> [5] "Blank_ch4_cyc8" "DAPI_ch1_cyc1" "Blank_ch2_cyc1" "Blank_ch3_cyc1"
#> [9] "Blank_ch4_cyc1" "DAPI_ch1_cyc2" "DAPI_ch1_cyc3" "Empty_ch2_cyc3"
#> [13] "DAPI_ch1_cyc4" "DAPI_ch1_cyc5" "DAPI_ch1_cyc6" "Empty_ch2_cyc6"
features(sm_tonsil)
#> [1] "CD3e" "CD21" "CD68" "Actin" "CD45RO"
#> [6] "HisH3p" "PanCK" "Ki67" "CD11c" "ECad"
#> [11] "CD4" "CD31" "Podoplanin" "CD107a" "CD20"
#> [16] "CD8"
## List per Region
embeddings(sm_tonsil, "spatial")
#> $TonsilA
#> x y
#> TonsilA.125182 891 1996
#> TonsilA.125238 935 1993
#> TonsilA.125254 34 1991
#> TonsilA.125325 18 1985
#> TonsilA.125326 997 1986
#> TonsilA.125608 13 1964
#> TonsilA.125729 879 1956
#> TonsilA.125925 9 1938
#> TonsilA.126062 1736 1928
#> TonsilA.126166 1750 1919
#> TonsilA.126199 955 1917
#> TonsilA.126219 902 1921
#> TonsilA.126569 963 1889
...
Representations(sm_tonsil, "spatial")
#> $TonsilA
#> Representation object 'spatial'
#> Dimensions: 'x', 'y'
#> 5810 cells
#>
#> $TonsilB
#> Representation object 'spatial'
#> Dimensions: 'x', 'y'
#> 10694 cellsIn the future, additional methods will be added to summon images and masks. The infrastructure for retrieving these files from Enable DB is under construction.
Behind the scenes, most SpatialMap functions work using these methods. .smapply for instance, first dispatches on the activeAnalysis, iterating, grouping, or combining samples (with a Formal Analysis). Then it runs a function to perform some computation. Then it updates the appropriate data slots, replaces the old Regions with the modified ones, and returns a modified SpatialMap.