SpatialMap Quickstart: Introduction and Feature Demo
Quickstart_guide.RmdNavigate vignettes
library(facil)
library(emconnect)
library(SpatialMap)
library(dplyr)
library(patchwork)
library(ggplot2)
library(ggthemes)SpatialMap Intro
Importing data
SpatialMap is a suite of tools for exploratory spatial biology data analysis built on some convenient data abstractions.
To get started quickly, let’s load a toy SpatialMap (sm_tonsil) object containing CODEX data.
sm_tonsil <- load_sm_data("tonsil")Object / class basics
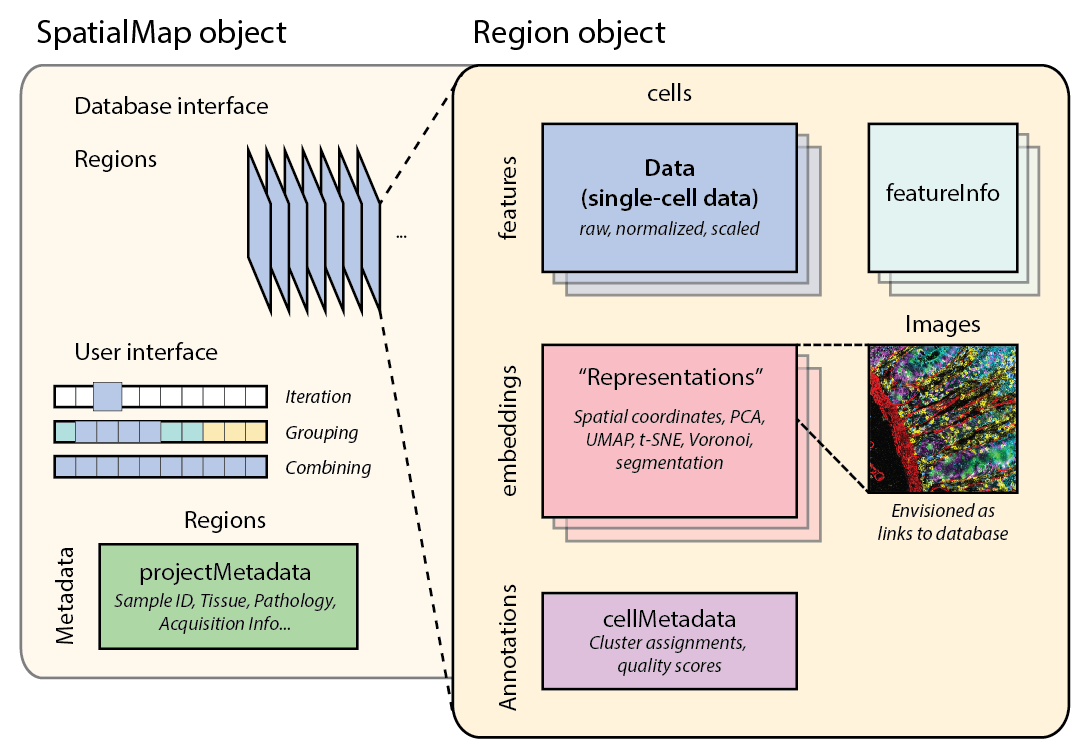
SpatialMap Schematic
The SpatialMap package is powered by abstractions (classes) that help represent and work with key aspects of spatial biology data. Those abstractions are: - SpatialMap - Region - Analysis - Representation - NN (nearest neighbors)
The package provides an interface such that users should primarily only need to interact with the main SpatialMap object. However, documentation for each of the other classes is provided, and more information about a particular object can be found using the str() method on the console.
SpatialMap
A SpatialMap object is a container that holds annotated spatial biology data. An object can be consist of multiple individual samples or acquisitions, which are called Regions. Regions can be combined as an Analysis. The SpatialMap object itself contains project information such as sample metadata, analysis settings, and feature information.
sm_tonsil
#> SpatialMap object
#> Active analysis: regions
#> + 16,504 total cells
#> + 16 features
#> + 2 Regions:
#> - TonsilA
#> - TonsilB
projectMetadata(sm_tonsil) %>% head()
#> color width n_channels height projection_visualizer_uuid
#> TonsilA grayscale 9408 4 9072 96e6af5a-f612-4f97-a294-c580dcd41915
#> TonsilB grayscale 9408 4 9072 f0b41218-209e-4946-9a1d-1fb7fb3e7a0d
#> sample_label n_levels assay_id n_cycles
#> TonsilA EnableShare 5 1 8
#> TonsilB EnableShare2 5 1 8
#> base_image_visualizer_uuid experiment_label axes
#> TonsilA 728d42b7-77ea-4319-a48a-7f7340bf182d enableshare TCYX
#> TonsilB 2007da53-7ede-492e-84b6-f2d2ab32a09d enableshare2 TCYX
#> assay_metadata assay_description
#> TonsilA {"image_mpp": 0.377, "num_amp_cycles": 0}
#> TonsilB {"image_mpp": 0.377, "num_amp_cycles": 0}
#> experiment_id visual_quality assay_name region_display_label
#> TonsilA 164 true CODEX EnableShare
#> TonsilB 355 true CODEX EnableShare2
#> biomarker_expression_version segmentation_version Region coverslip_id
#> TonsilA 1 1 TonsilA 164
#> TonsilB 1 1 TonsilB 164
#> study_name
#> TonsilA Enable Tonsil
#> TonsilB Enable Tonsil
cells(sm_tonsil) %>% head()
#> [1] "TonsilA.125182" "TonsilA.125238" "TonsilA.125254" "TonsilA.125325"
#> [5] "TonsilA.125326" "TonsilA.125608"
Regions(sm_tonsil)
#> [1] "TonsilA" "TonsilB"
showsettings(sm_tonsil)
#> Parameter Value
#> 1 cores 1
#> 2 parallel future
#> 3 future.initialized FALSE
#> 4 show.progress FALSE
#> 5 Rstudio TRUE
#> 6 python.path none
#> 7 python.environment spatialmap_py
#> 8 python.method reticulate
#> 9 platform unix
#> 10 local.images FALSE
#> 11 filtration soft
#> 12 normalization log
#> 13 max_cell.size 1600
#> 14 min_cell.size 100
#> 15 max_signal.sum 125000
#> 16 min_signal.sum 5000
#> 17 max_signal.cv 4
#> 18 min_signal.cv 1
#> 19 max_normalized.signal.sum 70
#> 20 min_normalized.signal.sum 10
#> 21 max_DNA 10000
#> 22 min_DNA 500
#> 23 max_num.markers 40
#> 24 min_num.markers 2
#> 25 max_dist.n 300
#> 26 max_dist.smooth 100
#> 27 exclude_partitions
#> 28 SpatialMap.version 0.5.20-->Repaired_0.5.25As an exercise, we can add to the projectMetadata using straightforward R conventions
new.pm <- projectMetadata(sm_tonsil)
new.pm[["sample_label"]] <- c("sample1", "sample2")
projectMetadata(sm_tonsil) <- new.pmRegion
A Region holds the expression data, cell coordinates, cell metadata, spatial analyses, and more. It is very similar to an anndata object in Python or a SingleCellExperiment object in R.
cellMetadata(sm_tonsil) %>% head()
#> cell_id cell.id anno316_some_categories anno916_Leiden_v1
#> TonsilA.125182 125182 TonsilA.125182 yay Cluster 2
#> TonsilA.125238 125238 TonsilA.125238 nay Cluster 2
#> TonsilA.125254 125254 TonsilA.125254 yay Cluster 1
#> TonsilA.125325 125325 TonsilA.125325 yay Cluster 1
#> TonsilA.125326 125326 TonsilA.125326 nay Cluster 6
#> TonsilA.125608 125608 TonsilA.125608 nay Cluster 1
#> anno248_test_subset_annotation_324
#> TonsilA.125182 p
#> TonsilA.125238 u
#> TonsilA.125254 t
#> TonsilA.125325 t
#> TonsilA.125326 b
#> TonsilA.125608 y
#> anno1189_uqc_test10_letters_qc_assoc anno1149_uqc_test10_letters
#> TonsilA.125182 b b
#> TonsilA.125238 b b
#> TonsilA.125254 b b
#> TonsilA.125325 c c
#> TonsilA.125326 c c
#> TonsilA.125608 b b
#> anno1133_uqc_test03_letters anno247_test_letters_324
#> TonsilA.125182 l p
#> TonsilA.125238 k u
#> TonsilA.125254 z t
#> TonsilA.125325 f t
#> TonsilA.125326 f b
#> TonsilA.125608 m y
#> anno1190_uqc_test10_letters_qc_assoc_postfilt
#> TonsilA.125182 <NA>
#> TonsilA.125238 <NA>
#> TonsilA.125254 b
#> TonsilA.125325 <NA>
#> TonsilA.125326 <NA>
#> TonsilA.125608 <NA>
#> anno253_Hierarchy.1 anno1797_uqc_test10_letters_221213
#> TonsilA.125182 not Th c
#> TonsilA.125238 not Th b
#> TonsilA.125254 Epithelial Cells b
#> TonsilA.125325 Epithelial Cells b
#> TonsilA.125326 Endothelial Cells b
#> TonsilA.125608 Epithelial Cells b
#> anno254_Neighborhood.clustering.based.on.annotation.set.253
#> TonsilA.125182 neighborhood_2
#> TonsilA.125238 neighborhood_2
#> TonsilA.125254 neighborhood_0
#> TonsilA.125325 neighborhood_0
#> TonsilA.125326 neighborhood_2
#> TonsilA.125608 neighborhood_0
#> anno1145_uqc_test08_letters anno1143_uqc_test08_letters Region
#> TonsilA.125182 l l TonsilA
#> TonsilA.125238 k k TonsilA
#> TonsilA.125254 z z TonsilA
#> TonsilA.125325 f f TonsilA
#> TonsilA.125326 f f TonsilA
#> TonsilA.125608 m m TonsilA
#> color width n_channels height
#> TonsilA.125182 grayscale 9408 4 9072
#> TonsilA.125238 grayscale 9408 4 9072
#> TonsilA.125254 grayscale 9408 4 9072
#> TonsilA.125325 grayscale 9408 4 9072
#> TonsilA.125326 grayscale 9408 4 9072
#> TonsilA.125608 grayscale 9408 4 9072
#> projection_visualizer_uuid sample_label n_levels
#> TonsilA.125182 96e6af5a-f612-4f97-a294-c580dcd41915 EnableShare 5
#> TonsilA.125238 96e6af5a-f612-4f97-a294-c580dcd41915 EnableShare 5
#> TonsilA.125254 96e6af5a-f612-4f97-a294-c580dcd41915 EnableShare 5
#> TonsilA.125325 96e6af5a-f612-4f97-a294-c580dcd41915 EnableShare 5
#> TonsilA.125326 96e6af5a-f612-4f97-a294-c580dcd41915 EnableShare 5
#> TonsilA.125608 96e6af5a-f612-4f97-a294-c580dcd41915 EnableShare 5
#> assay_id n_cycles base_image_visualizer_uuid
#> TonsilA.125182 1 8 728d42b7-77ea-4319-a48a-7f7340bf182d
#> TonsilA.125238 1 8 728d42b7-77ea-4319-a48a-7f7340bf182d
#> TonsilA.125254 1 8 728d42b7-77ea-4319-a48a-7f7340bf182d
#> TonsilA.125325 1 8 728d42b7-77ea-4319-a48a-7f7340bf182d
#> TonsilA.125326 1 8 728d42b7-77ea-4319-a48a-7f7340bf182d
#> TonsilA.125608 1 8 728d42b7-77ea-4319-a48a-7f7340bf182d
#> experiment_label segmentation_visualizer_uuid axes
#> TonsilA.125182 enableshare 9d6258e3-1532-40fb-9503-0831f8661358 TCYX
#> TonsilA.125238 enableshare 9d6258e3-1532-40fb-9503-0831f8661358 TCYX
#> TonsilA.125254 enableshare 9d6258e3-1532-40fb-9503-0831f8661358 TCYX
#> TonsilA.125325 enableshare 9d6258e3-1532-40fb-9503-0831f8661358 TCYX
#> TonsilA.125326 enableshare 9d6258e3-1532-40fb-9503-0831f8661358 TCYX
#> TonsilA.125608 enableshare 9d6258e3-1532-40fb-9503-0831f8661358 TCYX
#> assay_metadata assay_description
#> TonsilA.125182 {"image_mpp": 0.377, "num_amp_cycles": 0}
#> TonsilA.125238 {"image_mpp": 0.377, "num_amp_cycles": 0}
#> TonsilA.125254 {"image_mpp": 0.377, "num_amp_cycles": 0}
#> TonsilA.125325 {"image_mpp": 0.377, "num_amp_cycles": 0}
#> TonsilA.125326 {"image_mpp": 0.377, "num_amp_cycles": 0}
#> TonsilA.125608 {"image_mpp": 0.377, "num_amp_cycles": 0}
#> experiment_id visual_quality assay_name region_display_label
#> TonsilA.125182 164 true CODEX EnableShare
#> TonsilA.125238 164 true CODEX EnableShare
#> TonsilA.125254 164 true CODEX EnableShare
#> TonsilA.125325 164 true CODEX EnableShare
#> TonsilA.125326 164 true CODEX EnableShare
#> TonsilA.125608 164 true CODEX EnableShare
#> biomarker_expression_version segmentation_version study_name
#> TonsilA.125182 1 1 Enable Tonsil
#> TonsilA.125238 1 1 Enable Tonsil
#> TonsilA.125254 1 1 Enable Tonsil
#> TonsilA.125325 1 1 Enable Tonsil
#> TonsilA.125326 1 1 Enable Tonsil
#> TonsilA.125608 1 1 Enable Tonsil
featureInfo(sm_tonsil[[1]])
#> features
#> CD3e CD3e
#> CD21 CD21
#> CD68 CD68
#> Actin Actin
#> CD45RO CD45RO
#> HisH3p HisH3p
#> PanCK PanCK
#> Ki67 Ki67
#> CD11c CD11c
#> ECad ECad
#> CD4 CD4
#> CD31 CD31
#> Podoplanin Podoplanin
#> CD107a CD107a
#> CD20 CD20
#> CD8 CD8
# It's sometimes useful to annotate cells with sample-level metadata. We can do this with a quick function call.
# > This is done by default when pulling data from ATLAS
sm_tonsil <- mergeProjectMetadata(sm_tonsil)
cellMetadata(sm_tonsil) %>% head()
#> cell_id cell.id anno316_some_categories anno916_Leiden_v1
#> TonsilA.125182 125182 TonsilA.125182 yay Cluster 2
#> TonsilA.125238 125238 TonsilA.125238 nay Cluster 2
#> TonsilA.125254 125254 TonsilA.125254 yay Cluster 1
#> TonsilA.125325 125325 TonsilA.125325 yay Cluster 1
#> TonsilA.125326 125326 TonsilA.125326 nay Cluster 6
#> TonsilA.125608 125608 TonsilA.125608 nay Cluster 1
#> anno248_test_subset_annotation_324
#> TonsilA.125182 p
#> TonsilA.125238 u
#> TonsilA.125254 t
#> TonsilA.125325 t
#> TonsilA.125326 b
#> TonsilA.125608 y
#> anno1189_uqc_test10_letters_qc_assoc anno1149_uqc_test10_letters
#> TonsilA.125182 b b
#> TonsilA.125238 b b
#> TonsilA.125254 b b
#> TonsilA.125325 c c
#> TonsilA.125326 c c
#> TonsilA.125608 b b
#> anno1133_uqc_test03_letters anno247_test_letters_324
#> TonsilA.125182 l p
#> TonsilA.125238 k u
#> TonsilA.125254 z t
#> TonsilA.125325 f t
#> TonsilA.125326 f b
#> TonsilA.125608 m y
#> anno1190_uqc_test10_letters_qc_assoc_postfilt
#> TonsilA.125182 <NA>
#> TonsilA.125238 <NA>
#> TonsilA.125254 b
#> TonsilA.125325 <NA>
#> TonsilA.125326 <NA>
#> TonsilA.125608 <NA>
#> anno253_Hierarchy.1 anno1797_uqc_test10_letters_221213
#> TonsilA.125182 not Th c
#> TonsilA.125238 not Th b
#> TonsilA.125254 Epithelial Cells b
#> TonsilA.125325 Epithelial Cells b
#> TonsilA.125326 Endothelial Cells b
#> TonsilA.125608 Epithelial Cells b
#> anno254_Neighborhood.clustering.based.on.annotation.set.253
#> TonsilA.125182 neighborhood_2
#> TonsilA.125238 neighborhood_2
#> TonsilA.125254 neighborhood_0
#> TonsilA.125325 neighborhood_0
#> TonsilA.125326 neighborhood_2
#> TonsilA.125608 neighborhood_0
#> anno1145_uqc_test08_letters anno1143_uqc_test08_letters Region
#> TonsilA.125182 l l TonsilA
#> TonsilA.125238 k k TonsilA
#> TonsilA.125254 z z TonsilA
#> TonsilA.125325 f f TonsilA
#> TonsilA.125326 f f TonsilA
#> TonsilA.125608 m m TonsilA
#> color width n_channels height
#> TonsilA.125182 grayscale 9408 4 9072
#> TonsilA.125238 grayscale 9408 4 9072
#> TonsilA.125254 grayscale 9408 4 9072
#> TonsilA.125325 grayscale 9408 4 9072
#> TonsilA.125326 grayscale 9408 4 9072
#> TonsilA.125608 grayscale 9408 4 9072
#> projection_visualizer_uuid sample_label n_levels
#> TonsilA.125182 96e6af5a-f612-4f97-a294-c580dcd41915 EnableShare 5
#> TonsilA.125238 96e6af5a-f612-4f97-a294-c580dcd41915 EnableShare 5
#> TonsilA.125254 96e6af5a-f612-4f97-a294-c580dcd41915 EnableShare 5
#> TonsilA.125325 96e6af5a-f612-4f97-a294-c580dcd41915 EnableShare 5
#> TonsilA.125326 96e6af5a-f612-4f97-a294-c580dcd41915 EnableShare 5
#> TonsilA.125608 96e6af5a-f612-4f97-a294-c580dcd41915 EnableShare 5
#> assay_id n_cycles base_image_visualizer_uuid
#> TonsilA.125182 1 8 728d42b7-77ea-4319-a48a-7f7340bf182d
#> TonsilA.125238 1 8 728d42b7-77ea-4319-a48a-7f7340bf182d
#> TonsilA.125254 1 8 728d42b7-77ea-4319-a48a-7f7340bf182d
#> TonsilA.125325 1 8 728d42b7-77ea-4319-a48a-7f7340bf182d
#> TonsilA.125326 1 8 728d42b7-77ea-4319-a48a-7f7340bf182d
#> TonsilA.125608 1 8 728d42b7-77ea-4319-a48a-7f7340bf182d
#> experiment_label segmentation_visualizer_uuid axes
#> TonsilA.125182 enableshare 9d6258e3-1532-40fb-9503-0831f8661358 TCYX
#> TonsilA.125238 enableshare 9d6258e3-1532-40fb-9503-0831f8661358 TCYX
#> TonsilA.125254 enableshare 9d6258e3-1532-40fb-9503-0831f8661358 TCYX
#> TonsilA.125325 enableshare 9d6258e3-1532-40fb-9503-0831f8661358 TCYX
#> TonsilA.125326 enableshare 9d6258e3-1532-40fb-9503-0831f8661358 TCYX
#> TonsilA.125608 enableshare 9d6258e3-1532-40fb-9503-0831f8661358 TCYX
#> assay_metadata assay_description
#> TonsilA.125182 {"image_mpp": 0.377, "num_amp_cycles": 0}
#> TonsilA.125238 {"image_mpp": 0.377, "num_amp_cycles": 0}
#> TonsilA.125254 {"image_mpp": 0.377, "num_amp_cycles": 0}
#> TonsilA.125325 {"image_mpp": 0.377, "num_amp_cycles": 0}
#> TonsilA.125326 {"image_mpp": 0.377, "num_amp_cycles": 0}
#> TonsilA.125608 {"image_mpp": 0.377, "num_amp_cycles": 0}
#> experiment_id visual_quality assay_name region_display_label
#> TonsilA.125182 164 true CODEX EnableShare
#> TonsilA.125238 164 true CODEX EnableShare
#> TonsilA.125254 164 true CODEX EnableShare
#> TonsilA.125325 164 true CODEX EnableShare
#> TonsilA.125326 164 true CODEX EnableShare
#> TonsilA.125608 164 true CODEX EnableShare
#> biomarker_expression_version segmentation_version study_name
#> TonsilA.125182 1 1 Enable Tonsil
#> TonsilA.125238 1 1 Enable Tonsil
#> TonsilA.125254 1 1 Enable Tonsil
#> TonsilA.125325 1 1 Enable Tonsil
#> TonsilA.125326 1 1 Enable Tonsil
#> TonsilA.125608 1 1 Enable Tonsil
#> coverslip_id
#> TonsilA.125182 164
#> TonsilA.125238 164
#> TonsilA.125254 164
#> TonsilA.125325 164
#> TonsilA.125326 164
#> TonsilA.125608 164Regions can contain Representations, which can be accessed or modified with methods that “get” or “set”. By default, each Region will have a spatial representation, since it must be initialized with X/Y coordinates.
# Save the Region to its own object for now
reg <- sm_tonsil[[1]]
Representations(reg)
#> $spatial
#> Representation object 'spatial'
#> Dimensions: 'x', 'y'
#> 5810 cells
Representations(reg, "spatial")
#> Representation object 'spatial'
#> Dimensions: 'x', 'y'
#> 5810 cellsWe can plot different features over that spatial information easily.
p1 <- plotRepresentation(reg, "spatial", what = "panck", trim = c(0, .95))
p2 <- plotRepresentation(reg, "spatial", what = "cd8", trim = c(0, .95))
p1 + p2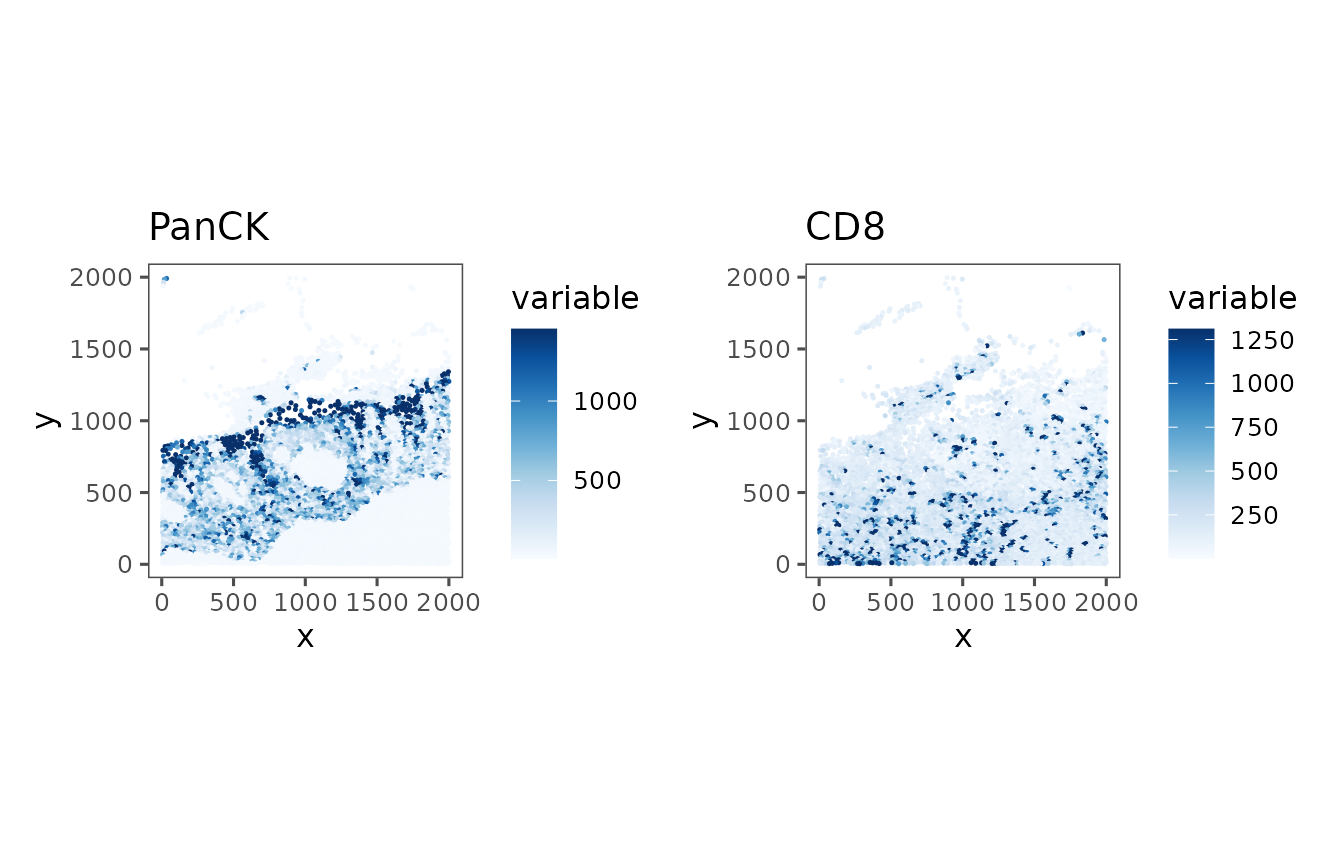
Basic Operations
SpatialMap and Region objects can be subsetted, often using base R syntax, such as [ and [[. We can also use special “setter” methods, such as the features<- function, to subset the features. In this case, features not included in the markers.i.like vector will be dropped. Careful though, as those markers can’t be retrieved from the object.
# Subset SpatialMaps
subset.sm_tonsil <- sm_tonsil[1]
# Subset Region cells
reg.subset <- sm_tonsil[[1]][1:4]
# Subset features
features(reg.subset)
#> [1] "CD3e" "CD21" "CD68" "Actin" "CD45RO"
#> [6] "HisH3p" "PanCK" "Ki67" "CD11c" "ECad"
#> [11] "CD4" "CD31" "Podoplanin" "CD107a" "CD20"
#> [16] "CD8"
markers.i.like <- c("PanCK", "CD11c", "CD20", "CD8", "Ki67")
features(reg.subset) <- markers.i.like
features(reg.subset)
#> [1] "PanCK" "CD11c" "CD20" "CD8" "Ki67"Creating your own functions
Central to SpatialMap is the idea that the Regions that comprise the object can be modified either as individual samples or in combination. The .smapply() is a powerful function, analagous to R’s apply, that allows users to write their own functions on the SpatialMap object.
Below, we write an anonymous function that subsets each Region to a specific x-y range.
small_tonsil <- .smapply(object = sm_tonsil, function(r) {
## Retreive spatial embeddings from Region 'r'
emb <- embeddings(r, "spatial")
## Take the desired subset from the top corner
my.subset <- emb[, "x"] < 1000 & inv(emb[, "y"]) < 1000
## Check that some cells remain (otherwise things could break)
if (sum(my.subset) < 2) {
stop("Subset is empty")
}
## Subset Region r and return
r[my.subset]
})
dim(reg)
#> cells features
#> 5810 16
dim(small_tonsil[[1]])
#> cells features
#> 371 16Data Quality Control and Filtering
Next, we will run functions to calculate basic QC metrics and pass cells through QC filters. The results can be visualized using special plotting functions.
dna <- "DAPI"
sm_tonsil <- sm_tonsil %>%
QCMetrics(DNA = dna) %>%
FilterCodex()
#> Calculating quality control metrics...
#>
#> Filtering cells...
#>
qc.list <- plotQCSummary(sm_tonsil, group.variable = "sample_label")
wrap_plots(qc.list)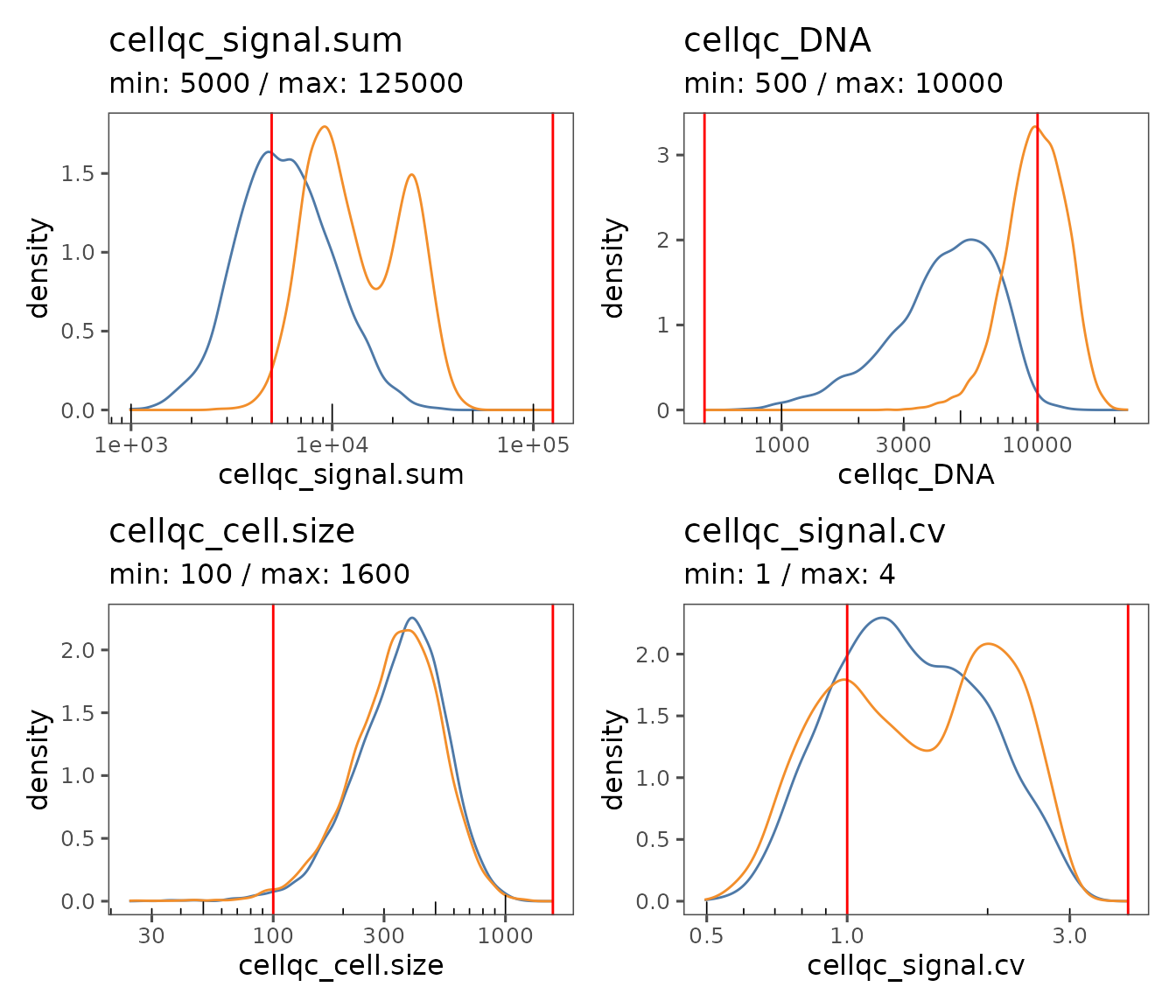
It may be apparent that the QC thresholds used were not perfect. We can easily modify them by updating the object settings, and re-running the QC and Filter commands.
Filter results are stored in cellMetadata, and can be plotted using plotFilteredCells, which is simply a special case of the plotRepresentation function.
showsettings(sm_tonsil)
#> Parameter Value
#> 1 cores 1
#> 2 parallel future
#> 3 future.initialized FALSE
#> 4 show.progress FALSE
#> 5 Rstudio TRUE
#> 6 python.path none
#> 7 python.environment spatialmap_py
#> 8 python.method reticulate
#> 9 platform unix
#> 10 local.images FALSE
#> 11 filtration soft
#> 12 normalization log
#> 13 max_cell.size 1600
#> 14 min_cell.size 100
#> 15 max_signal.sum 125000
#> 16 min_signal.sum 5000
#> 17 max_signal.cv 4
#> 18 min_signal.cv 1
#> 19 max_normalized.signal.sum 70
#> 20 min_normalized.signal.sum 10
#> 21 max_DNA 10000
#> 22 min_DNA 500
#> 23 max_num.markers 40
#> 24 min_num.markers 2
#> 25 max_dist.n 300
#> 26 max_dist.smooth 100
#> 27 exclude_partitions
#> 28 SpatialMap.version 0.5.20-->Repaired_0.5.25
new.settings <- c(
min_signal.cv = 0.75,
max_signal.cv = 3.5,
max_DNA = 20000,
min_DNA = 250,
min_signal.sum = 2500,
max_signal.sum = 100000
)
settings(sm_tonsil, names(new.settings)) <- new.settings
sm_tonsil <- sm_tonsil %>%
QCMetrics(DNA = dna) %>%
FilterCodex()
#> Calculating quality control metrics...
#>
#> Filtering cells...
#>
new.fs.list <- plotFilterSummary(sm_tonsil, group.variable = "Region")
wrap_plots(new.fs.list)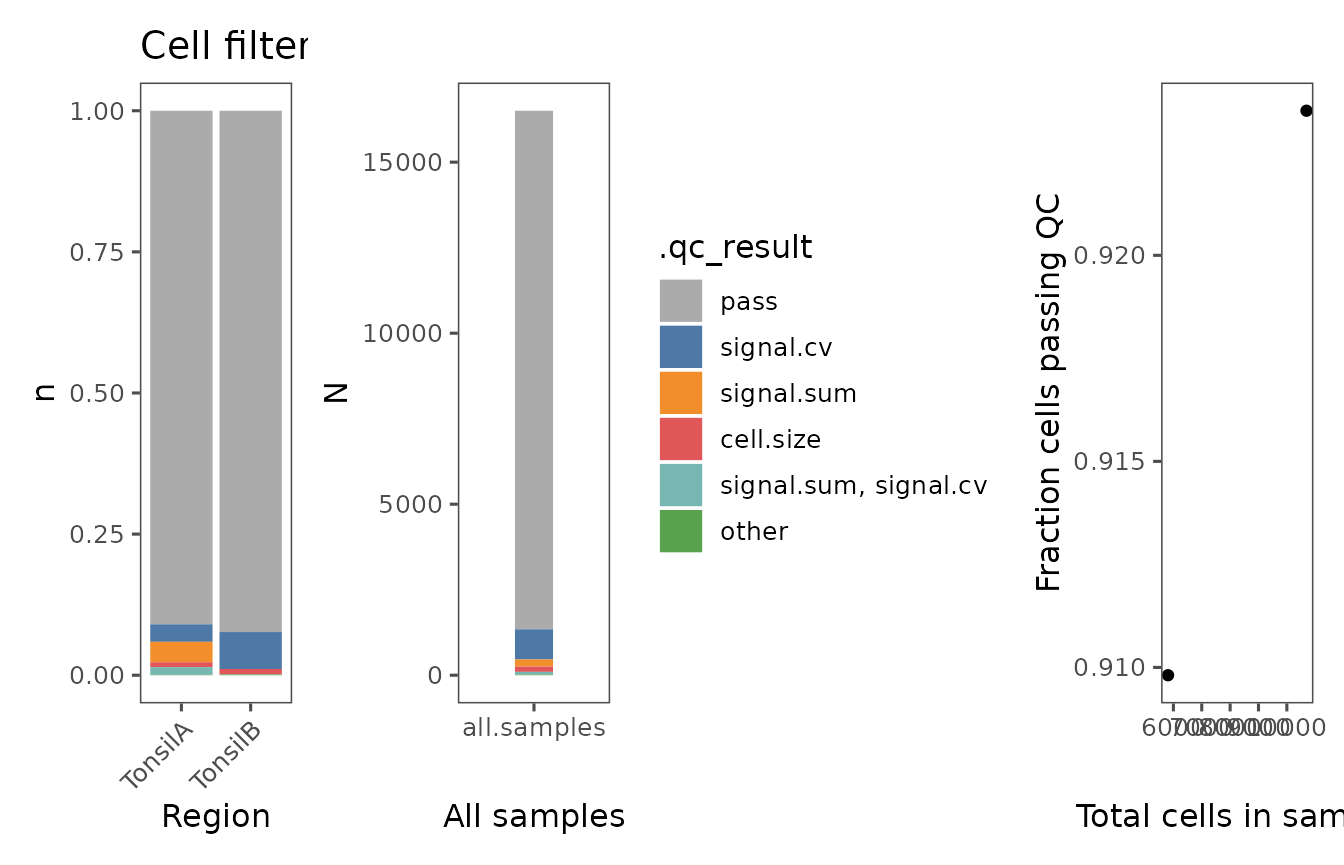
filter.plots <- plotFilteredCells(sm_tonsil)
filter.plots[[1]]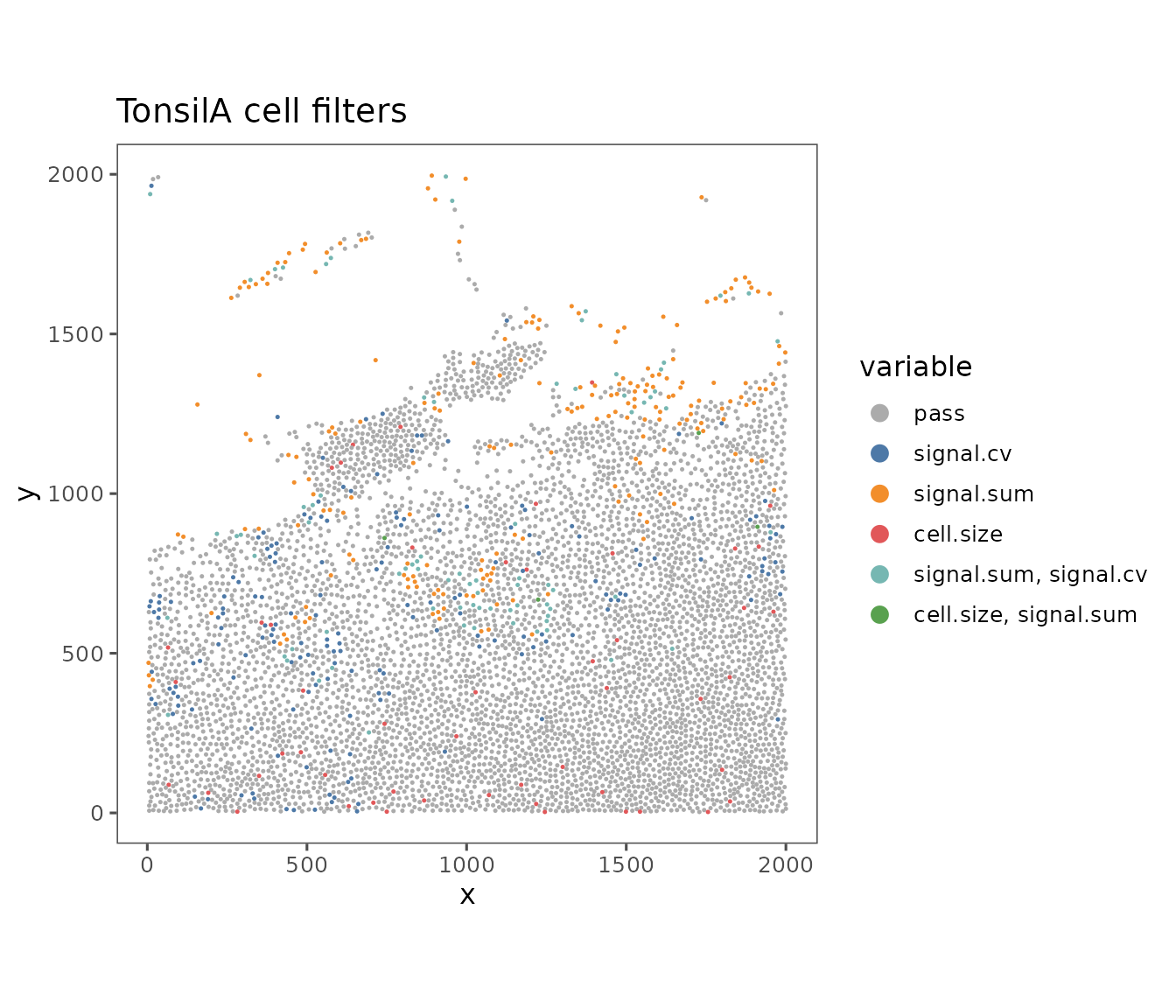
Finally, we may wish to label the filtered cells, or actually remove them entirely from our analysis. The filter function provides a parameter to choose. Note that the removed cells are not preserved anywhere after this step!
sm_tonsil <- FilterCodex(sm_tonsil, remove.filtered.cells = T)Data Normalization and Scaling
SpatialMap implements a select list of scaling and normalization functions curated for spatial proteomics data. Some simple transformations - arcsin transform and robust scaling - are shown below, and users are encouraged to explore the full suite in the Normalize documentation in (?Normalize) in order to find the workflows that best account for batch effects and experiments in their data.
sm_tonsil <- sm_tonsil %>%
Normalize(method = "asinh", from = "Data", to = "NormalizedData") %>%
Normalize(method = "scale", from = "NormalizedData", to = "ScaledData")
#> Normalizing data using method `asinh`
#>
#> Normalizing data using method `scale`
#>
featureInfo(sm_tonsil[[1]])
#> features 0% 10% 20% 30% 40% 50%
#> CD3e CD3e 13.600 31.7162 35.9138 39.5297 44.1938 49.6145
#> CD21 CD21 57.529 266.8636 339.8682 414.3092 496.8962 605.8785
#> CD68 CD68 43.103 167.8457 191.2824 212.5469 233.0440 259.7695
#> Actin Actin 19.737 36.5922 39.7528 41.9627 43.8038 45.6465
#> CD45RO CD45RO 19.584 150.2757 174.6478 196.1867 218.5066 242.2745
#> HisH3p HisH3p 9.064 48.1388 54.7516 60.1876 65.4418 70.6130
#> PanCK PanCK 8.109 28.6548 32.7278 37.4576 45.3874 64.8410
#> Ki67 Ki67 3.420 92.9732 119.7084 141.2547 164.7920 195.4330
#> CD11c CD11c 34.033 82.7721 92.9756 101.8474 110.9640 120.0590
#> ECad ECad 0.794 137.4073 191.1620 243.4006 293.1336 358.0260
#> CD4 CD4 14.184 50.2858 59.9098 70.1698 81.9488 96.2305
#> CD31 CD31 8.864 31.7347 35.4242 38.5848 41.4254 44.4345
#> Podoplanin Podoplanin 2.376 111.0212 151.6054 199.5184 326.0710 560.9920
#> CD107a CD107a 26.656 91.2956 125.0510 155.6178 190.9350 225.7370
#> CD20 CD20 22.632 135.6811 268.5706 391.3278 504.6348 612.6625
#> CD8 CD8 0.568 68.9498 97.0338 120.0068 140.4384 162.9525
#> 60% 70% 80% 90% 100%
#> CD3e 58.7494 75.7193 112.4684 206.1743 1022.907
#> CD21 753.1586 967.7830 1287.6744 1934.0145 15034.270
#> CD68 290.6922 327.3372 385.2952 511.9733 10383.041
#> Actin 47.5920 49.9096 52.9030 57.7335 129.109
#> CD45RO 271.2812 310.7763 362.5008 473.4737 1925.049
#> HisH3p 76.7472 83.9486 95.2082 117.8481 8850.312
#> PanCK 178.2786 339.3262 532.5656 914.7127 7299.914
#> Ki67 237.3502 292.4716 419.2380 996.3913 22920.352
#> CD11c 131.1460 145.3691 167.6772 211.4985 1189.828
#> ECad 440.6974 600.2093 908.2984 1550.6641 6958.132
#> CD4 115.0546 143.9397 191.7948 287.8391 4679.053
#> CD31 48.5814 54.2942 65.1392 95.7470 875.383
#> Podoplanin 1048.1634 1894.0597 3400.1404 5627.5859 29556.791
#> CD107a 263.8120 305.9724 357.9064 443.3744 2948.113
#> CD20 716.9250 826.6439 942.0000 1100.5661 5771.062
#> CD8 193.4120 234.7125 311.4080 649.5986 12148.939
#> arcsinh_scale_factors
#> CD3e 179.569
#> CD21 1699.341
#> CD68 956.412
#> Actin 198.764
#> CD45RO 873.239
#> HisH3p 273.758
#> PanCK 163.639
#> Ki67 598.542
#> CD11c 464.878
#> ECad 955.810
#> CD4 299.549
#> CD31 177.121
#> Podoplanin 758.027
#> CD107a 625.255
#> CD20 1342.853
#> CD8 485.169
m1 <- "CD8"
par(mfrow = c(1, 3))
r1 <- sm_tonsil[[1]]
hist(Data(r1, "Data")[m1, ])
hist(Data(r1, "NormalizedData")[m1, ])
hist(Data(r1, "ScaledData")[m1, ])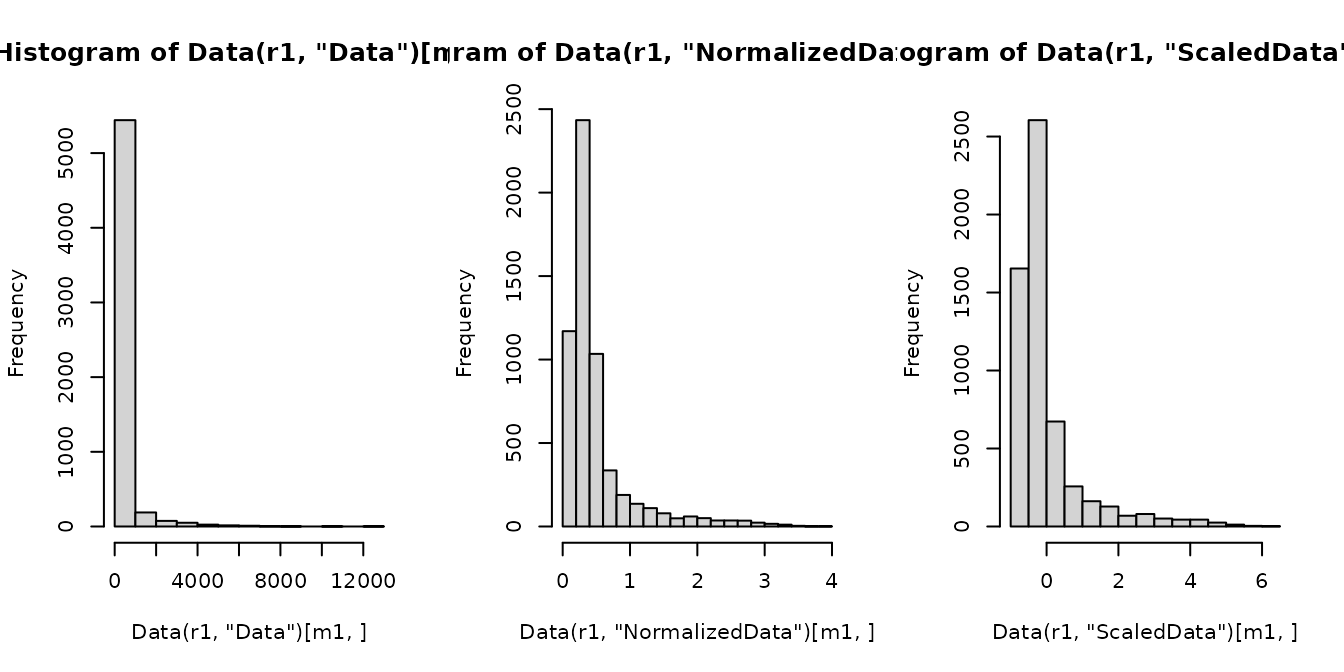
Clustering and Dimensionality Reduction
The Analysis Class
An Analysis is a group of Regions. When an Analysis is active, SpatialMap functions will automatically combine the relevant Region data and operate on the combined data. Otherwise, SpatialMap functions will operate on each Region separately. This logic is all handled through the .smapply() function, which we first discussed above.
Analyses can contain their own Representation objects (for example, PC representations or UMAPs), nearest neighbor graphs, cells, and features.
Here, we activate a new analysis containing both of our Regions in order to run unsupervised clustering on our entire expression data.
sm_tonsil <- createAnalysis(sm_tonsil, id = "combined", regions = Regions(sm_tonsil))
#> New analysis created.
#> 2 Regions
#> 16504 cells
#> 16 common features
#>
#> Added Analysis `combined` to SpatialMap project 'SpatialMap Project'.
#> Setting as active Analysis
#>
sm_tonsil <-
runUMAP(sm_tonsil,
data.slot = "ScaledData",
PCA = TRUE,
dims = 8,
n_neighbors = 50,
verbose = TRUE
)
#> By analysis: 'combined'
#>
#> Computing PCA
#>
#> Computing UMAP
#>
#> 16:04:19 UMAP embedding parameters a = 0.9922 b = 1.112
#> 16:04:19 Read 16504 rows and found 8 numeric columns
#> 16:04:20 Building Annoy index with metric = cosine, n_trees = 50
#> 0% 10 20 30 40 50 60 70 80 90 100%
#> [----|----|----|----|----|----|----|----|----|----|
#> **************************************************|
#> 16:04:22 Writing NN index file to temp file /tmp/RtmpHX1Hgq/file106f664b164d
#> 16:04:22 Searching Annoy index using 2 threads, search_k = 5000
#> 16:04:27 Annoy recall = 100%
#> 16:04:28 Commencing smooth kNN distance calibration using 2 threads with target n_neighbors = 50
#> 16:04:29 Initializing from normalized Laplacian + noise (using irlba)
#> 16:04:30 Commencing optimization for 200 epochs, with 1070580 positive edges
#> 16:04:38 Optimization finished
sm_tonsil <- clusterCells(sm_tonsil, method = "leiden")
#> By analysis: 'combined'
#> Plotting Representations
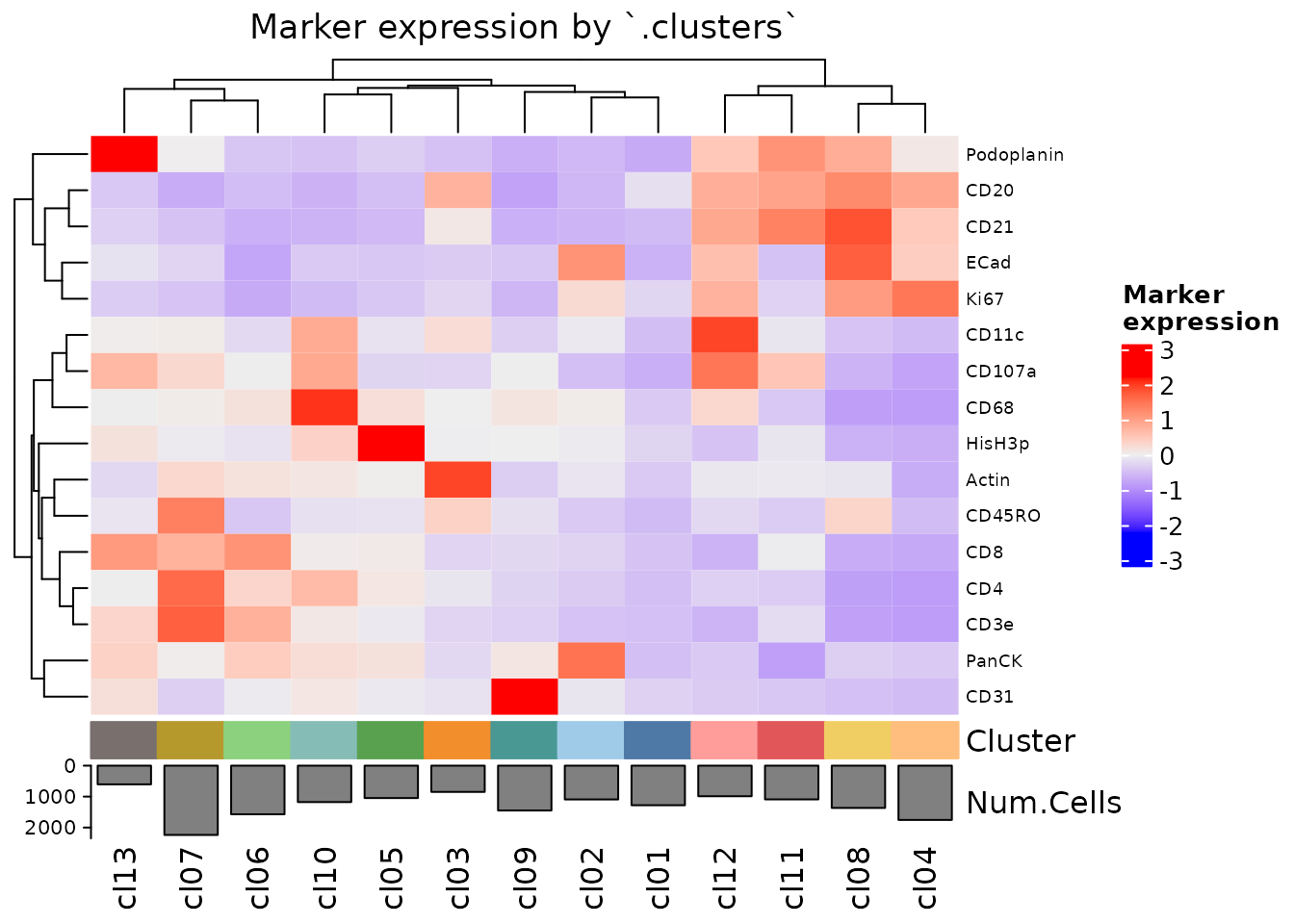
Our UMAP and PCA results can be plotted with the same code as the spatial plots: plotRepresentation(). We can also now plot a summarized heatmap of expression values across clusters.
plotExpressionHeatmap(sm_tonsil, data.slot = "ScaledData", summarize.across = ".clusters", scaling = "none")
#> Plotting expression from slot 'ScaledData'.
#> Scaling = 'none'
#> By analysis: 'combined'
#> 
pu1 <- plotRepresentation(sm_tonsil, "umap", ".clusters")
#> By analysis: 'combined'
#>
pu2 <- plotRepresentation(sm_tonsil, "umap", "Region")
#> By analysis: 'combined'
#>
pu1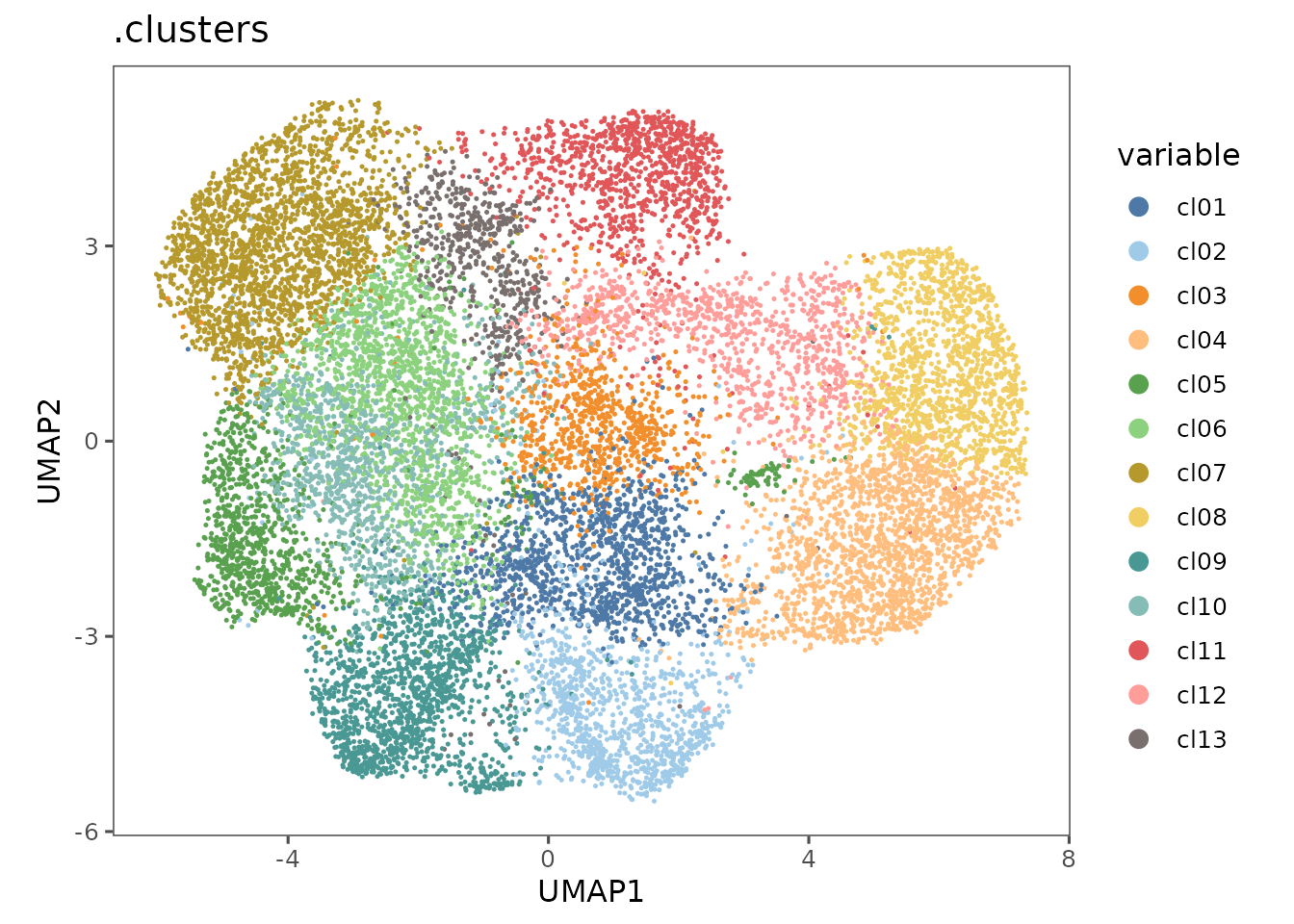
pu2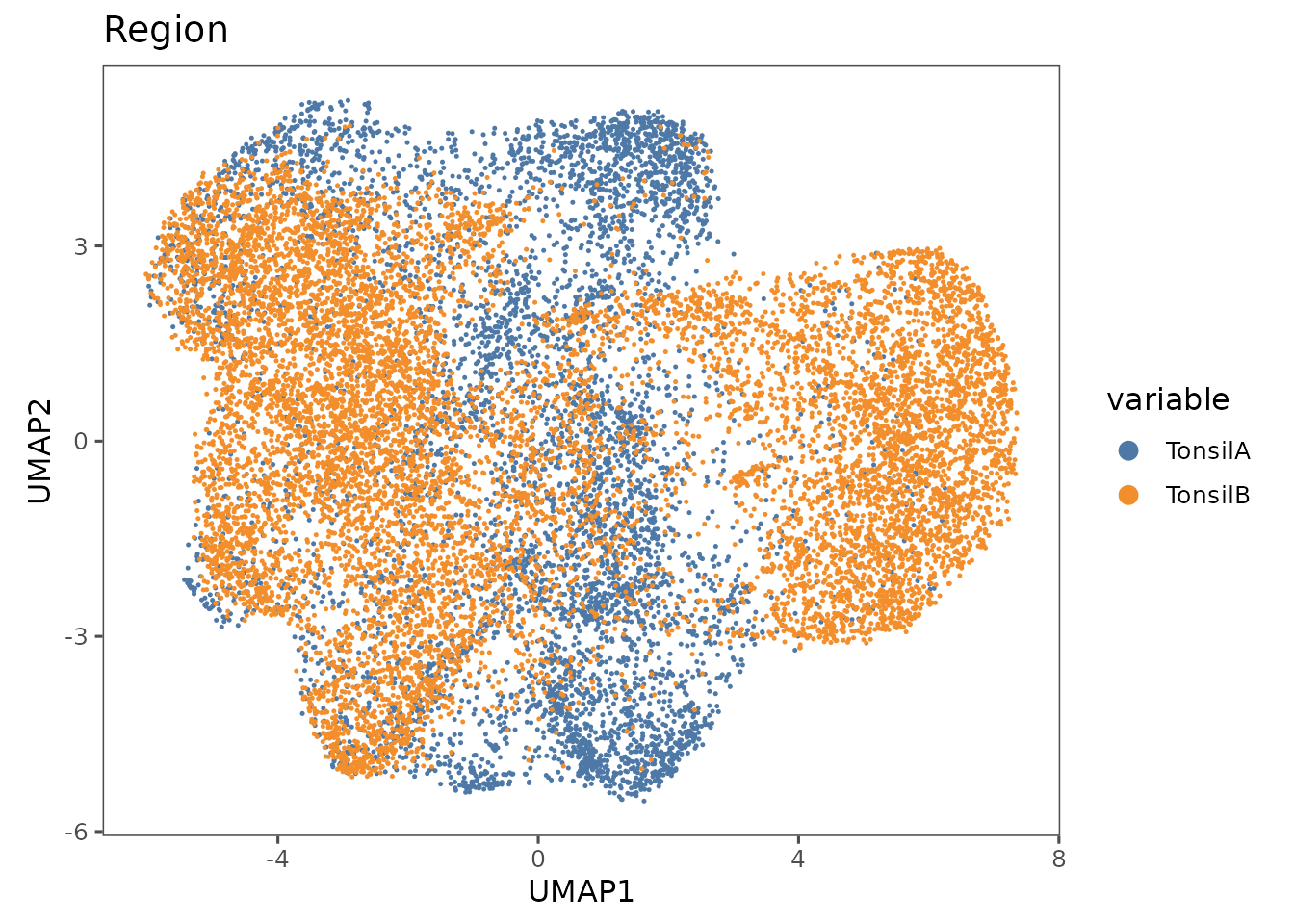
Another common use here is to plot marker expression in the reduced dimensions:
ue1 <- plotRepresentation(sm_tonsil, "umap", "panck", data.slot = "ScaledData")
#> By analysis: 'combined'
#>
ue2 <- plotRepresentation(sm_tonsil, "umap", "cd8", data.slot = "ScaledData")
#> By analysis: 'combined'
#>
ue1 + ue2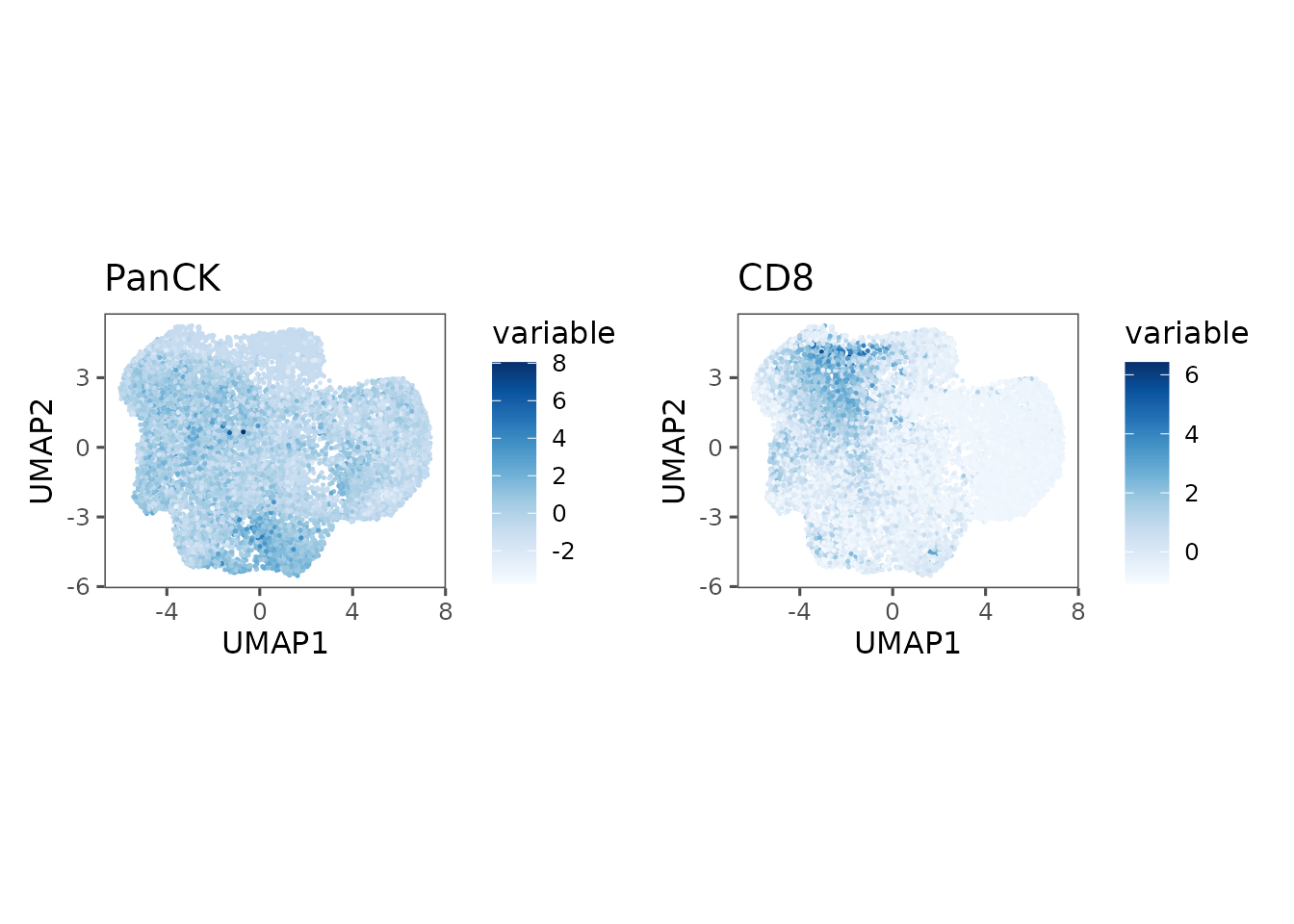
Handling Metadata
Having viewed our expression values by cluster, we might want to annotate them manually. We can use base R to construct a named vector mapping cluster labels to their manual annotations. Then, SpatialMap provides a helper function to map the automatic values to the new ones. Finally, we can add those values to the object in the cellMetadata slot using addCellMetadata()
# Renaming clusters
cluster.labels <- c(
cl01 = "B Cell",
cl02 = "T Cell"
)
old.labels <- cellMetadata(sm_tonsil)$.clusters
new.labels <- mapValues(old.labels, cluster.labels)
#> Warning: mapping returned 13926 NA values
sm_tonsil <- addCellMetadata(sm_tonsil, metadata = new.labels, col.names = "Cell.type")Spatial Analysis
Of course, the most interesting insights from CODEX or other spatial data should be from the spatial information. Here, we demo two methods for spatial analysis. First, pairwise adjacency, which computes how much more likely two cell groups are to interact in space than you would expect by chance (taking into account the graph structure and cell group proportions of a sample).
Statistical analysis of cell neighborhoods in the different samples
activeAnalysis(sm_tonsil) <- "regions"
sm_tonsil <- spatialNearestNeighbors(sm_tonsil, representation = "spatial", method = "knn", k = 5)
sm_tonsil <- pairwiseAdjacency(sm_tonsil, nn = "spatial_knn_5", feature = ".clusters")
pa.plots <- list()
pa.plots[["enrichment"]] <- plotPairwiseAdjacency(
sm_tonsil,
nn = "spatial_knn_5",
feature = ".clusters",
what = "log10.padj.enrichment",
cluster_rows = F,
cluster_columns = F
)
pa.plots[["depletion"]] <- plotPairwiseAdjacency(
sm_tonsil,
nn = "spatial_knn_5",
feature = ".clusters",
what = "log10.padj.depletion",
cluster_rows = F,
cluster_columns = F
)We can plot those results:
pa.plots$enrichment[[1]]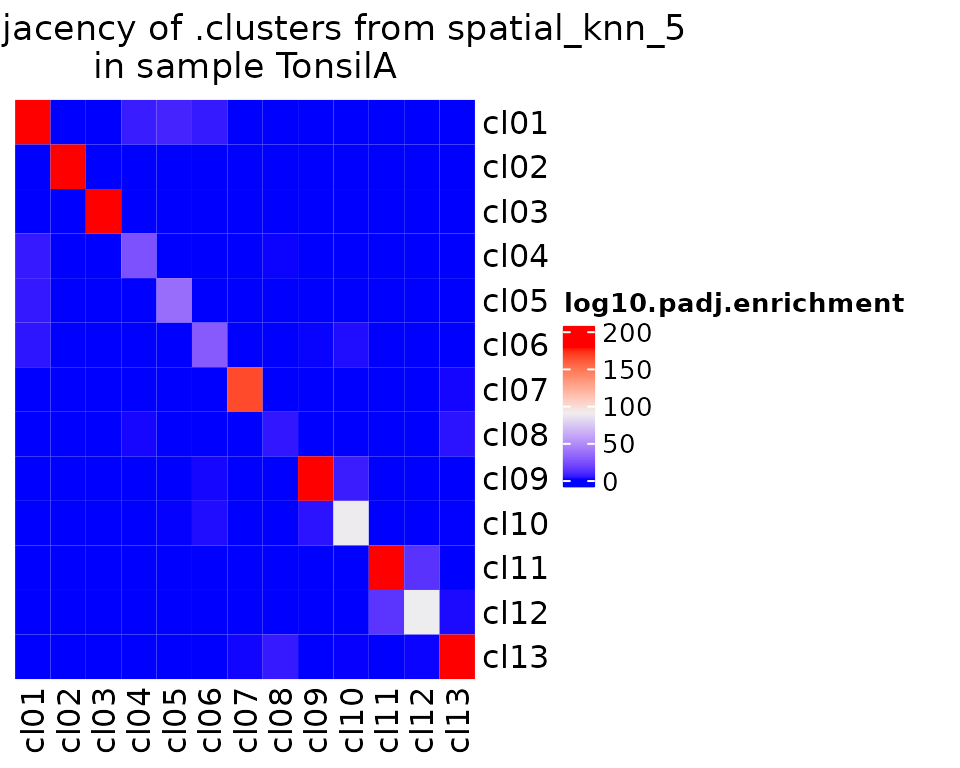
pa.plots$depletion[[1]]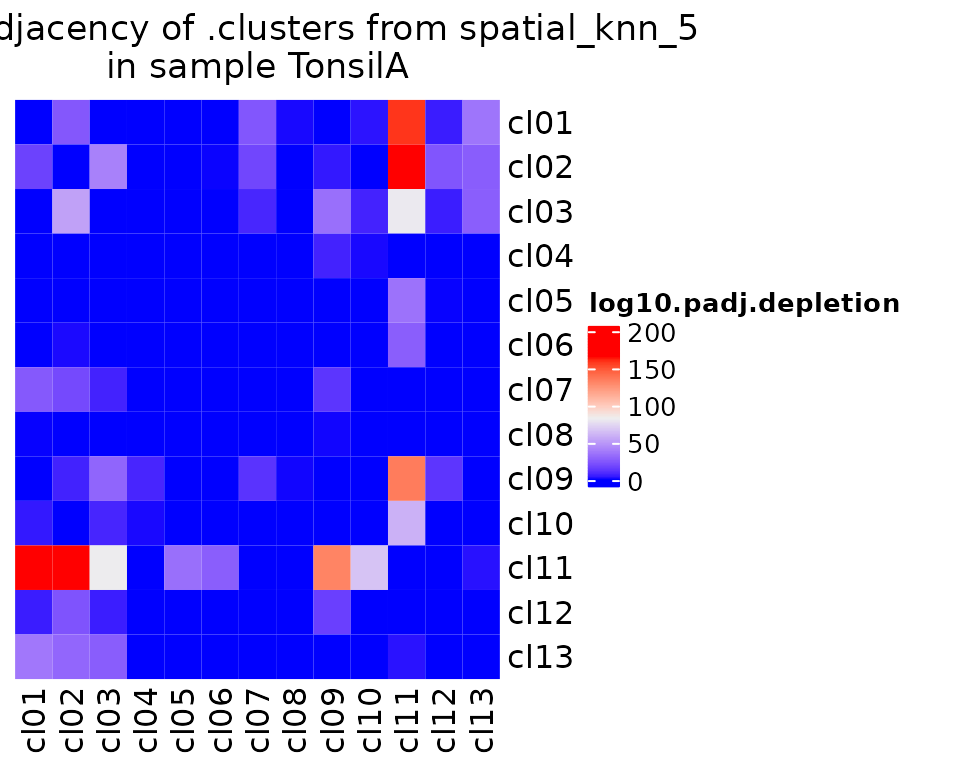
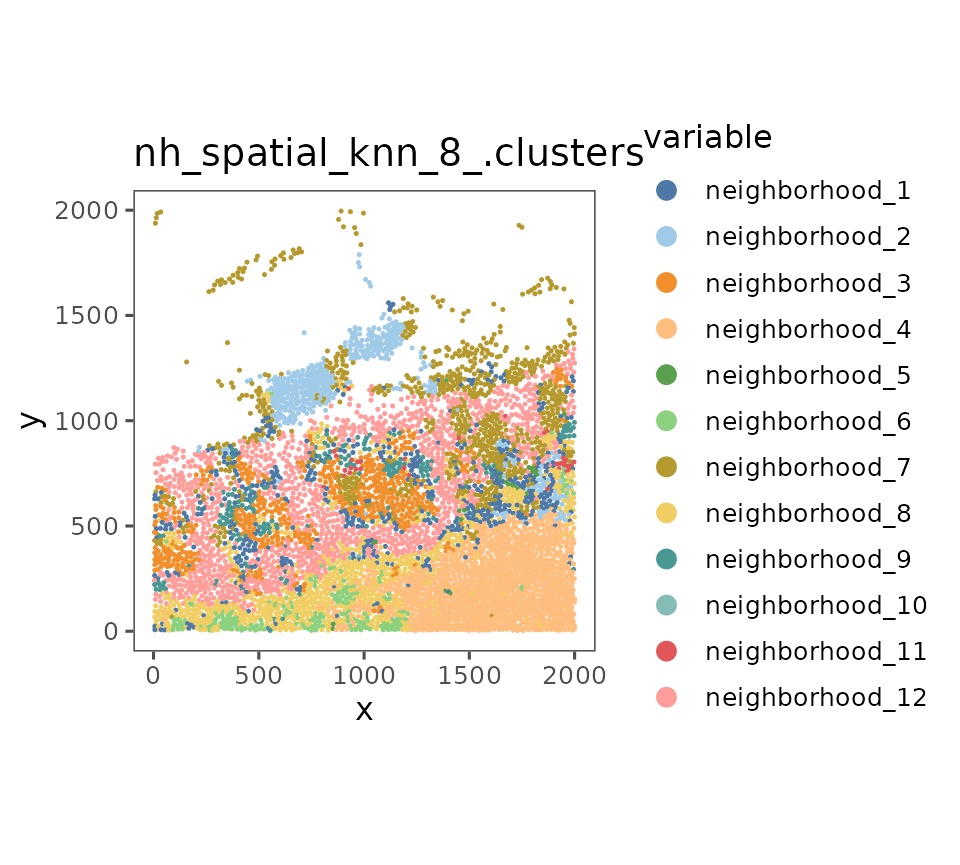
Second, neighborhood analysis, which was described in Schurch et al 2020. Essentially, a nearest neighbor network is computed, the identities of each cell’s neighbors are tallied up, and those vectors are then clustered across the whole dataset. The resulting clusters can be called “neighborhoods” because they describe the overall identity of “windows” of cells.
sm_tonsil <- sm_tonsil %>%
spatialNearestNeighbors(k = 8) %>%
cellNeighborhoods(nn = "spatial_knn_8", feature = ".clusters")
sm_tonsil <- neighborhoodClusters(sm_tonsil, nn = "spatial_knn_8", feature = ".clusters")
#> Warning: did not converge in 10 iterations
#> Warning: did not converge in 10 iterations
print(head(cellMetadata(sm_tonsil)))
#> cell_id cell.id anno316_some_categories anno916_Leiden_v1
#> TonsilA.125182 125182 TonsilA.125182 yay Cluster 2
#> TonsilA.125238 125238 TonsilA.125238 nay Cluster 2
#> TonsilA.125254 125254 TonsilA.125254 yay Cluster 1
#> TonsilA.125325 125325 TonsilA.125325 yay Cluster 1
#> TonsilA.125326 125326 TonsilA.125326 nay Cluster 6
#> TonsilA.125608 125608 TonsilA.125608 nay Cluster 1
#> anno248_test_subset_annotation_324
#> TonsilA.125182 p
#> TonsilA.125238 u
#> TonsilA.125254 t
#> TonsilA.125325 t
#> TonsilA.125326 b
#> TonsilA.125608 y
#> anno1189_uqc_test10_letters_qc_assoc anno1149_uqc_test10_letters
#> TonsilA.125182 b b
#> TonsilA.125238 b b
#> TonsilA.125254 b b
#> TonsilA.125325 c c
#> TonsilA.125326 c c
#> TonsilA.125608 b b
#> anno1133_uqc_test03_letters anno247_test_letters_324
#> TonsilA.125182 l p
#> TonsilA.125238 k u
#> TonsilA.125254 z t
#> TonsilA.125325 f t
#> TonsilA.125326 f b
#> TonsilA.125608 m y
#> anno1190_uqc_test10_letters_qc_assoc_postfilt
#> TonsilA.125182 <NA>
#> TonsilA.125238 <NA>
#> TonsilA.125254 b
#> TonsilA.125325 <NA>
#> TonsilA.125326 <NA>
#> TonsilA.125608 <NA>
#> anno253_Hierarchy.1 anno1797_uqc_test10_letters_221213
#> TonsilA.125182 not Th c
#> TonsilA.125238 not Th b
#> TonsilA.125254 Epithelial Cells b
#> TonsilA.125325 Epithelial Cells b
#> TonsilA.125326 Endothelial Cells b
#> TonsilA.125608 Epithelial Cells b
#> anno254_Neighborhood.clustering.based.on.annotation.set.253
#> TonsilA.125182 neighborhood_2
#> TonsilA.125238 neighborhood_2
#> TonsilA.125254 neighborhood_0
#> TonsilA.125325 neighborhood_0
#> TonsilA.125326 neighborhood_2
#> TonsilA.125608 neighborhood_0
#> anno1145_uqc_test08_letters anno1143_uqc_test08_letters Region
#> TonsilA.125182 l l TonsilA
#> TonsilA.125238 k k TonsilA
#> TonsilA.125254 z z TonsilA
#> TonsilA.125325 f f TonsilA
#> TonsilA.125326 f f TonsilA
#> TonsilA.125608 m m TonsilA
#> color width n_channels height
#> TonsilA.125182 grayscale 9408 4 9072
#> TonsilA.125238 grayscale 9408 4 9072
#> TonsilA.125254 grayscale 9408 4 9072
#> TonsilA.125325 grayscale 9408 4 9072
#> TonsilA.125326 grayscale 9408 4 9072
#> TonsilA.125608 grayscale 9408 4 9072
#> projection_visualizer_uuid sample_label n_levels
#> TonsilA.125182 96e6af5a-f612-4f97-a294-c580dcd41915 EnableShare 5
#> TonsilA.125238 96e6af5a-f612-4f97-a294-c580dcd41915 EnableShare 5
#> TonsilA.125254 96e6af5a-f612-4f97-a294-c580dcd41915 EnableShare 5
#> TonsilA.125325 96e6af5a-f612-4f97-a294-c580dcd41915 EnableShare 5
#> TonsilA.125326 96e6af5a-f612-4f97-a294-c580dcd41915 EnableShare 5
#> TonsilA.125608 96e6af5a-f612-4f97-a294-c580dcd41915 EnableShare 5
#> assay_id n_cycles base_image_visualizer_uuid
#> TonsilA.125182 1 8 728d42b7-77ea-4319-a48a-7f7340bf182d
#> TonsilA.125238 1 8 728d42b7-77ea-4319-a48a-7f7340bf182d
#> TonsilA.125254 1 8 728d42b7-77ea-4319-a48a-7f7340bf182d
#> TonsilA.125325 1 8 728d42b7-77ea-4319-a48a-7f7340bf182d
#> TonsilA.125326 1 8 728d42b7-77ea-4319-a48a-7f7340bf182d
#> TonsilA.125608 1 8 728d42b7-77ea-4319-a48a-7f7340bf182d
#> experiment_label segmentation_visualizer_uuid axes
#> TonsilA.125182 enableshare 9d6258e3-1532-40fb-9503-0831f8661358 TCYX
#> TonsilA.125238 enableshare 9d6258e3-1532-40fb-9503-0831f8661358 TCYX
#> TonsilA.125254 enableshare 9d6258e3-1532-40fb-9503-0831f8661358 TCYX
#> TonsilA.125325 enableshare 9d6258e3-1532-40fb-9503-0831f8661358 TCYX
#> TonsilA.125326 enableshare 9d6258e3-1532-40fb-9503-0831f8661358 TCYX
#> TonsilA.125608 enableshare 9d6258e3-1532-40fb-9503-0831f8661358 TCYX
#> assay_metadata assay_description
#> TonsilA.125182 {"image_mpp": 0.377, "num_amp_cycles": 0}
#> TonsilA.125238 {"image_mpp": 0.377, "num_amp_cycles": 0}
#> TonsilA.125254 {"image_mpp": 0.377, "num_amp_cycles": 0}
#> TonsilA.125325 {"image_mpp": 0.377, "num_amp_cycles": 0}
#> TonsilA.125326 {"image_mpp": 0.377, "num_amp_cycles": 0}
#> TonsilA.125608 {"image_mpp": 0.377, "num_amp_cycles": 0}
#> experiment_id visual_quality assay_name region_display_label
#> TonsilA.125182 164 true CODEX EnableShare
#> TonsilA.125238 164 true CODEX EnableShare
#> TonsilA.125254 164 true CODEX EnableShare
#> TonsilA.125325 164 true CODEX EnableShare
#> TonsilA.125326 164 true CODEX EnableShare
#> TonsilA.125608 164 true CODEX EnableShare
#> biomarker_expression_version segmentation_version study_name
#> TonsilA.125182 1 1 Enable Tonsil
#> TonsilA.125238 1 1 Enable Tonsil
#> TonsilA.125254 1 1 Enable Tonsil
#> TonsilA.125325 1 1 Enable Tonsil
#> TonsilA.125326 1 1 Enable Tonsil
#> TonsilA.125608 1 1 Enable Tonsil
#> coverslip_id cellqc_signal.sum cellqc_signal.mean
#> TonsilA.125182 164 2150.384 134.3990
#> TonsilA.125238 164 1643.021 102.6888
#> TonsilA.125254 164 5008.782 313.0489
#> TonsilA.125325 164 4780.682 298.7926
#> TonsilA.125326 164 2061.935 128.8709
#> TonsilA.125608 164 2562.896 160.1810
#> cellqc_signal.min cellqc_signal.max cellqc_signal.median
#> TonsilA.125182 25.351 616.531 71.2760
#> TonsilA.125238 22.278 264.544 94.6020
#> TonsilA.125254 43.392 1176.123 186.8460
#> TonsilA.125325 43.724 1091.923 216.6215
#> TonsilA.125326 26.698 333.908 84.0285
#> TonsilA.125608 27.512 415.596 135.3460
#> cellqc_signal.sd cellqc_signal.vars cellqc_DNA cellqc_cell.size
#> TonsilA.125182 156.32565 24437.708 2711.271 484
#> TonsilA.125238 75.59541 5714.666 1606.299 763
#> TonsilA.125254 358.27940 128364.126 1644.679 357
#> TonsilA.125325 304.16653 92517.280 1650.913 417
#> TonsilA.125326 99.92029 9984.064 2454.006 520
#> TonsilA.125608 116.45369 13561.462 2145.376 651
#> cellqc_signal.cv .pass_qc .qc_result .clusters
#> TonsilA.125182 1.1631459 FALSE signal.sum cl01
#> TonsilA.125238 0.7361601 FALSE signal.sum, signal.cv cl01
#> TonsilA.125254 1.1444839 TRUE pass cl02
#> TonsilA.125325 1.0179854 TRUE pass cl03
#> TonsilA.125326 0.7753516 FALSE signal.sum cl04
#> TonsilA.125608 0.7270131 FALSE signal.cv cl01
#> Cell.type nh_spatial_knn_8_.clusters
#> TonsilA.125182 B Cell neighborhood_10
#> TonsilA.125238 B Cell neighborhood_10
#> TonsilA.125254 T Cell neighborhood_10
#> TonsilA.125325 <NA> neighborhood_10
#> TonsilA.125326 <NA> neighborhood_10
#> TonsilA.125608 B Cell neighborhood_10
plotRepresentation(sm_tonsil[[1]], "spatial", "nh_spatial_knn_8_.clusters")